Dokument badawczy na temat genetyki człowieka (10031 słów)
Oto Twój artykuł badawczy na temat genetyki człowieka, chromosomów i genów!
Dziedziczymy pewne fizyczne i biochemiczne postacie naszych rodziców i przodków. Przekazywanie odziedziczonych postaci lub cech przez pokolenia jest znane jako dziedziczność. Genetyka jest tą gałęzią nauk biologicznych, która zajmuje się badaniem podstawowych zasad dziedziczności.

Zdjęcie dzięki uprzejmości: mdsalaries.com/wp-content/uploads/2011/11/shutterstock_61775431.jpg
Ustalono, że dziedziczne cechy lub cechy są przekazywane przez geny chromosomów. Wyrażanie odziedziczonych postaci jest jednak modyfikowane przez środowiska, w których człowiek się rozwija i rozwija.
Podstawowa znajomość ludzkich chromosomów i genów jest zatem niezbędna do zrozumienia zasad genetyki.
Chromosomy:
Chromosomy są głęboko zabarwionymi strukturami nitkowatymi w jądrze każdej komórki zwierzęcej. Geny są przenoszone przez chromosomy w liniowych seriach jako części specyficznej cząsteczki DNA. Poszczególne chromosomy są widoczne pod mikroskopem tylko podczas podziału komórki.
Podczas interfazy komórki jądro zawiera sieć nici lub granulek chromatyny, ale nie indywidualny chromosom, ponieważ każdy chromosom zostaje rozwinięty w długą cienką nitkę, która jest poza rozdzielczością mikroskopu świetlnego. Ale niektóre chromosomy pozostają zwinięte w miejscach i są identyfikowane jako granule chromatyny w interfazie (ryc. 11-1).

Odwijana część chromosomu jest znana jako euchromatyna, która jest aktywna genetycznie; zwinięta część nazywana jest heterochromatyną, która jest genetycznie obojętna. Podczas podziału komórki każdy chromosom jest ciasno zwinięty na całej jego długości i staje się krótszy i grubszy. Ostatecznie poszczególne chromosomy są łatwo widoczne pod mikroskopem (ryc. 11-2).

Dlatego chromosomy są genetycznie nieaktywne podczas podziału komórki. Wszystkie biochemiczne aktywności chromosomów w postaci replikacji DNA, tworzenia mRNA i syntezy białek zachodzą podczas interfazy, która składa się z trzech etapów cyklu komórkowego - etapy G1 (Gap 1), S (synteza), G 2 (Gap 2) . Replikacja DNA odbywa się w etapie S i obejmuje okres około 7 godzin.
Każdy chromosom przedstawia pierwotne zwężenie znane jako centromer lub kinetocore, który jest przyłączony do wrzeciona achromatycznego podczas podziału komórki i organizuje tworzenie mikrotubuli chromosomowej (Fig. 11-3a).

W profazie podziału komórki, każdy chromo-some dzieli się wzdłużnie na dwie chromatydy, z wyjątkiem centromeru (ryc. 1 l-3b). Wolne końce chromatydów są znane jako telomery, które w stanie nienaruszonym nie pozwalają na fuzję z chromatydami sąsiadujących chromosomów. Chromatydy niektórych chromosomów wykazują wtórne kontrakty w pobliżu jednego końca, a segment chromatyd dalej od zwężenia tworzy bryły satelitów (ryc. 11-3c, d). Uważa się, że wtórne zwężenia organizują tworzenie jąder.
Rodzaje chromosomów (ryc. 11 -4):
Centromery zajmują pozycje zmienne w stosunku do ich pary chromatyd. Odpowiednio, chromosomy można nazwać metacentrycznymi, gdy centromer jest w środku, sub-metacentryczny, gdy centromer jest nieznacznie przesunięty ze środka, akrocentryczny, gdy znajduje się blisko końca, i telocentryczny, jeśli centromer zajmuje koniec chromosomu. Telosomatyczne chromosomy nie są obecne w człowieku, chyba że patologiczne. Większość chromosomów akrocentrycznych wykazuje ciała satelitów na krótszych ramionach oddzielonych wtórnymi przewężeniami. Krótsze ramiona chromatyd są symbolizowane przez p i dłuższe ramiona przez q.

Liczba chromosomów:
Liczba chromosomów jest stała u gatunku. U ludzi liczba ta wynosi 46 (diploidalna) we wszystkich komórkach somatycznych i niedojrzałych komórkach zarodkowych, ale liczba 23 (haploidalna) w dojrzałych komórkach zarodkowych lub gametach. Dokładna liczba 46 chromosomów w każdej komórce somatycznej zdrowego człowieka została po raz pierwszy wykryta przez Tjio i Levan (1956) wraz z pojawieniem się kultury tkankowej.
Niektóre zaburzenia dziedziczne są związane ze zmianą liczby chromosomów. Kiedy liczba ta jest zwiększona o wiele haploidalnych (23) chromosomów (innych niż liczba diploidalna), schorzenie to nazywa się poliploidią. Jeśli poliploidy, np. Triploidia lub tetraploidia, wpływają na wszystkie komórki somatyczne, wskaźnik przeżywalności jest niski. Polyploidalna w stadium zygoty może być wynikiem zapłodnienia komórki jajowej przez więcej niż jedno plemniki.
W normalnych warunkach poliploidię można znaleźć w niektórych komórkach wątroby i błony śluzowej pęcherza moczowego. Może się to zdarzyć w telofazie mitozy, gdy po utworzeniu dwóch błon jądrowych otaczających diploidalną liczbę chromosomów, cytoplazma nie jest dzielona, a dwie błony jądrowe łączą się w podwójną liczbę diploidalnych chromosomów.
Aneuploidia to stan, w którym liczba chromosomów jest zmieniana o jeden lub więcej, ale nie przez wielokrotność haploidów. Większość zagrożeń związanych z liczbą chromosomów ma miejsce w anafazie. Po rozdzieleniu centromerów jeden lub więcej chromosomów nie migruje prawidłowo z powodu nieprawidłowej funkcji wrzeciona achromatycznego. Zjawisko to jest znane jako niedozwolone.
W rezultacie obaj członkowie konkretnej pary idą do jednej komórki potomnej, która otrzymuje dodatkowy chromosom (trisomia), a druga komórka potomna ma niedobór tego chromosomu (monosomia). Czasami po rozszczepieniu centromeru jeden członek nowo utworzonego chromosomu oddziela się jak zwykle tworząc normalny dopełniacz chromosomów w jednej komórce potomnej, podczas gdy drugi członek nie osiąga przeciwnego bieguna wrzeciona powodując niedobór tego chromosomu (monosomia) w drugim komórka córki. Jest to tak zwane opóźnienie anafazowe.
Brak dysocjacji może mieć miejsce w mitozy lub mejozie i może obejmować chromosomy płciowe, a także autosomy. Autosomalny brak dysocjacji jest mniej żywotny, szczególnie gdy wpływa na duże chromosomy. Nasze ciało jest bardziej szkodliwe dla komórek trisomicznych niż monosomowe. Komórki monosomowe degenerują się wcześnie. Zespół Turnera z kobietą z 45 chromosomową konstytucją XO jest prawdopodobnie jedynym przykładem zdolnego do życia monosomowego osobnika. Jeżeli w pierwszym odszczepieniu zygoty następuje niedyspozycja, wówczas wszystkie komórki są aneuploidalne, a osobnik wykazuje mozaikę, przy czym połowa wszystkich komórek jest trisomiczna, a druga połowa monosomowa.
Gdy w mejozie I nie występuje dysocjacja, wszystkie cztery gamety są nieprawidłowe (dwa z 24 chromosomami i dwa z 22 chromosomami). Jeśli ma to miejsce w mejozie II, dwie gameta są normalne i dwie nienormalne. Gdy zapłodnienie zachodzi pomiędzy normalnymi i nieprawidłowymi gametami, wszystkie komórki organizmu pochodzące z tej zygoty są aneuploidami. Nisisjunkcja w gametogenezie jest czasami obserwowana u starszych kobiet (35 lat i więcej). Prawdopodobnie pierwotny oocyt, który rozpoczyna pierwszy podział mejotyczny w życiu prenatalnym, kończy proces tuż przed owulacją po dłuższej przerwie o około 40 lat dłużej. Opóźnione zakończenie pierwszej mejozy oocytów może faworyzować niedysjunkcję.
Układy chromosomów:
Czterdzieści dwa chromosomy w każdej komórce somatycznej normalnego człowieka są rozmieszczone w 23 parach. Dwadzieścia dwie pary są znane jako autosomy, których geny regulują postacie ciała; pozostała para jest znana jako chromosomy płci, które regulują głównie cechy płciowe. Jeden członek z każdej pary jest ojcem, a drugi z matczynego pochodzenia.
Parowanie odbywa się między identycznymi chromosomami, które mają identyczną długość, pozycję centromeru, wzór pasmowy i rozkład genów. Sparowane chromosomy są znane jako homologiczne chromosomy (ryc. 11-5).

U samic chromosomy dwóch płci mają identyczną długość i są symbolizowane przez XX. U mężczyzn, sparowane chromosomy płciowe mają nierówną długość i są symbolizowane przez XY. Im dłuższy jest reprezentowany przez X, a krótszy przez Y. Podczas parowania chromosomów męskiej płci, oba mają części homologiczne i nie-ho-mologiczne (ryc. 11-6).

Geny lub cistrony, które są częścią specyficznej cząsteczki DNA, są zawarte w chromosomach w szeregu liniowym. Tworzą one funkcjonalne jednostki dziedzicznych postaci. Pozycja genu w chromosomie nazywa się jego locus, który jest wymieniony w odniesieniu do centromeru.
Geny nie zmieniają loci, z wyjątkiem zmian w morfologii chromosomów lub rekombinacji spowodowanej krzyżowaniem w mejozie. Geny zajmujące identyczne loci w parze homologicznych chromosomów są znane jako allelomorfy lub allele (patrz Fig. 11-5). Geny alleliczne regulują różne specyficzne cechy fizyczne i biochemiczne, poprzez tworzenie RNA i biosyntezę białek.
W przygotowaniu chromosomów z mitotycznej hodowli komórkowej (po zatrzymaniu podziału komórki w metafazie) homologiczne pary chromosomów nie są wizualizowane. Pary homologiczne są dopasowane tylko podczas kariotypowania z powiększonych fotomikrografii. Jednak w stadium zygotenu w profazie pierwszego podziału mejotycznego, homologiczne chromosomy znajdują się w parach ustanawiających zależność punkt-punkt; Zjawisko to znane jest jako synapsis.
Ciała Sex Chromatin lub Barr:
Podczas interfazy komórka somatyczna normalnej samicy przedstawia heterochromatynowo-wypukłe ciało poniżej błony jądrowej. Jest to znane jako chromatyna płciowa lub ciało Barr. Zostało to po raz pierwszy wykryte przez Barra i Bertrama w 1949 r. W jądrach komórek nerwu przeponowego u kotów. Z dwóch chromosomów X u normalnej samicy jeden z nich jest bardzo zwinięty, a drugi bardzo rozwinięty. Bardzo zwinięty genetycznie nieaktywny chromosom X tworzy ciało Barra, które jest otynkowane pod błoną jądrową (ryc. 11-7).

Ciała te pomagają w nuklearnym określaniu płci tkanek. Ciała Barr można łatwo znaleźć w tych komórkach, które mają jądra o otwartych twarzach. Zwykle ciała Barra są badane z komórek wymazu policzkowego lub poprzez obserwowanie ciał "pałeczek perkusyjnych" przyczepionych do jądra polimorficznych jądrowych leukocytów.
Liczba ciał Barr w komórce jest równa całkowitej liczbie chromosomów X minus jeden. U normalnej samicy z dwoma chromosomami X liczba osobników to jeden. W potrójnym X sydromie (XXX) liczba ta jest zwiększana do dwóch; u kobiet z zespołem Turnera mających tylko jeden chromosom X (XO), ciało Barr jest nieobecne. U mężczyzn z zespołem Klinefeltera z chromosomami XXY (trisomia) obecne jest ciało Barr.
Obecność chromosomu Y u mężczyzny jest wykrywana jako ciało intensywnie fluorescencyjne (ciało F) w jądrze, gdy rozmaz policzkowy jest wybarwiony barwnikiem flurochromowym i badany pod mikroskopem fluorescencyjnym. Ponieważ technika ta jest kosztowna, a szkiełko szybko się pogarsza, zwykle nie stosuje się jej do badania statusu chromatyny płci.
Struktura chemiczna chromosomów:
W analizie chemicznej stwierdzono, że każdy chromosom zawiera DNA, niewielką ilość RNA, histon i białka niehistonowe oraz jony metali. DNA jest najważniejszym i stabilnym molekularnym składnikiem chromosomów.
Ostatnie badania wykazały, że każdy chromosom eukariotyczny zawiera pojedynczą ciągłą dwuniciową cząsteczkę DNA. Większość cząsteczki DNA istnieje w chromosomie jako wysoce zwinięta lub złożona struktura. DNA w stanie aktywnej transkrypcji jest najbardziej rozszerzony i staje się euchromatyczny; nieaktywny region DNA pozostaje wysoce zwinięty i staje się heterochromatyczny. Stopień zwinięcia DNA zmienia się w zależności od szybkości syntezy białka w różnych fazach cyklu komórkowego.
W ludzkich chromosomach obserwuje się dwa typy stałych regionów heterochromatycznych;
(a) Fakultatywna heterochromatyna wpływa na nieaktywny chromosom X normalnej samicy. We wczesnej embriogenezie kobiety oba chromosomy X są aktywnie zaangażowane w rozwój jajników; następnie jeden z chromosomów X staje się trwale nieaktywny i tworzy heterochromatyczne ciało Barr.
(b) Konstytucyjna heterochromatyna jest obserwowana w pierwotnym i wtórnym przewężeniu chromosomów. Powtarzająca się sekwencja zasad DNA, bogatych w guaninę i cytozynę, jest obecna w konstytutywnej heterochromatynie i w ciałach satelitarnych. Powtarzające się DNA w niektórych częściach chromosomów prawdopodobnie koduje cząsteczki wewnętrzne w postaci rybosomalnego RNA, transferu RNA i białek regulatorowych.
Histony to podstawowe białka bogate w argininę i lizynę. Białka te są agregowane jako sferoidalne cząstki wzdłuż nici DNA, które są zwinięte wokół każdej cząstki i tworzą złożony organizm zwany nukleosomem lub v-ciałem (ryc. 11-8). Każdy nukleosom składa się z czterech par histonów ułożonych w dwie symetryczne grupy. Dowody eksperymentalne sugerują, że połączenie DNA z histonem tłumi aktywność genu.

Niehistonowe białka są kwaśne i tworzą wiele enzymów, np. Polimerazę DNA i polimerazę RNA. Niektóre z niehistonowych białek uwalniają histony od aktywności genów nukleosomu i derepresji.
Procedura analizy chromosomów:
Do badań cytogenetycznych chromosomów wybiera się komórki, które szybko się rozwijają i dzielą w kulturze. Najczęściej stosowanymi tkankami są skóra, szpik kostny i krew obwodowa.
Zasady przygotowania chromosomów z krwi obwodowej są następujące:
(a) Około 1-2 ml. krwi jest pobierana z żyły, heparynizowana i traktowana fitobigaglutyniną, ekstrahowana z czerwonej fasoli.
Fitohemaglutynina (PHA) stymuluje limfocyty (szczególnie komórki T) do proliferacji przez mitozę i selektywnie umożliwia aglutynację i sedymentację dojrzałych erytrocytów.
(b) Porcja osocza z zawieszonymi limfocytami jest teraz przenoszona do butelek hodowlanych w sterylnych warunkach zawierających TC199 (Difco) jako pożywkę hodowlaną. Inkubacja w butelce hodowlanej trwa około 3 dni w 37 ° C z dodatkiem streptomycyny i penicyliny jako konserwantów.
(c) Kolchicynę dodano do hodowli i trzymano przez około 2 godziny. Kolchicyna zatrzymuje podział komórkowy na metafazę, zapobiegając tworzeniu mikrotubul achromatycznego wrzeciona. W metafazie chromatydy zjednoczone przez centromery są maksymalnie skurczone.
(d) Komórki zbiera się przez odwirowanie zawartości butelki hodowlanej. Hipotoniczny roztwór cytrynianu sodu jest dodawany do komórek i inkubowany przez około 20 minut. Roztwór hipotoniczny umożliwia komórkom pęcznienie i zdyspergowanie chromosomów.
(e) Podłoże hipotoniczne odrzuca się przez odwirowanie. Teraz utrwalacze mieszaniny etanolu i kwasu octowego dodaje się do grudek komórek i delikatnie wytrząsa się z wytworzeniem zawiesiny komórkowej.
(f) Małe krople zawiesiny komórek umieszcza się na jednym końcu chemicznie oczyszczonych szkiełek. Suwaki pozostawiono do wyschnięcia w temperaturze pokojowej.
(g) Barwienie - Dla konwencjonalnego badania wzoru chromosomalnego, barwienie Giemsy jest szeroko stosowane z dobrymi wynikami (Fig. 11-9).

Precyzyjna identyfikacja poszczególnych chromosomów jest teraz możliwa dzięki odnotowaniu wzoru pasm na chromosomach po zastosowaniu dowolnej z czterech różnych technik barwienia:
(i) Pasma Q:
Kiedy chromosomy o ustalonej metafazie są wybarwione chlorowodorkiem chinakryny lub musztardą chinakrynową, niektóre prążki chromosomu pojawiają się jako obszary fluorescencyjne w mikroskopie fluorescencyjnym. Te wzory prążków Q (fluorescencyjne) są unikalne dla każdego chromosomu. Przypuszczalnie regiony pasm Q są bogatsze w bazy DNA adeniny (A) i tyminy (T) niż regiony międzypasmowe. Szczególnie duży prążek Q jest widoczny w dalszej części długiego ramienia chromosomu Y, nawet podczas interfazy.
(ii) G-banding:
Ustalone chromosomy są poddawane łagodnemu działaniu enzymów proteolitycznych (trypsyny) przed barwieniem. Enzymy są zdolne do denaturacji białka w chromosomach. Po zabarwieniu Giemsa po takim traktowaniu, wzór ciemnych prążków barwiących może być widoczny na chromosomach pod mikroskopem świetlnym.
Regiony chromosomów G i pasma Q ściśle odpowiadają sobie. Comings (1974) zasugerował, że białka pozostające po denaturacji mogą uniemożliwić dostawanie się materiału barwiącego do pewnych regionów DNA. Możliwe, że mniej białka może być związane z DNA bogatym w AT; to wyjaśnia zgodność pasm G i Q.
Pasy G i regiony pasma Q są bogate w pary zasad AT; odpowiadają one regionom heterochromatynowym chromosomów, w których replikacja DNA zachodzi nieco później. Regiony międzypasmowe są bogate w pary zasad GC.
(iii) wiązanie R:
Jest to odwrotność wiązania G, gdzie regiony pasmowe są demonstrowane przez barwienie Giemsa po ogrzaniu do 87 ° C. Wiązanie R jest komplementarne do wiązania G.
(iv) C-banding:
Po ostrym potraktowaniu utrwalonych chromosomów za pomocą alkaliów, kwasu lub soli, barwienie Giemsa ujawnia zabarwiony region, pasmo С, blisko centromeru. Opaska C nie jest jednak widoczna w chromosomie Y.
Pasy chromosomowe pomagają zlokalizować pewne nieprawidłowości w budowie chromosomu, takie jak delecja i translokacja określonych regionów chromosomów.
Karyotype:
Jest to proces uporządkowania chromosomów w porządku. Powiększona fotomikrografia "spreadu" chromosomu pochodzi z wybarwionego slajdu. Poszczególne chromosomy są wycinane ze zdjęcia, dopasowane do par homologicznych i są ułożone w sekwencji, najdłuższe chromosomy są umieszczone na początku, a najkrótsze na końcu.
Poszczególne chromosomy są identyfikowane na podstawie ich długości, położenia centromeru, stosunku długości ramion i obecności ciał satelitów na ramionach. (Ryc. 11-10) Wzorzec pasmowy dodatkowo przyczynia się do identyfikacji poszczególnych chromosomów. (Ryc. 11-11).


Klasyfikacja ludzkich chromosomów:
Zgodnie z klasyfikacją Denver System (1960), ludzkie chromosomy, w tym chromosomy płci, są ułożone w siedem grup od A do G, w kolejności malejącej długości.
(1) Grupa A:
Obejmuje pary 1, 2, 3 chromosomów. Każda z nich jest długa i metacentryczna. Jednakże chromosom 2 umieszczony w grupie A jest najdłuższym chromosomem sub-metacentrycznym.
(2) Grupa В:
Składa się z par 4 i 5 chromosomów, które są dość długie z centromerami submetacentrycznymi.
(3) Grupa С:
Jest to duża grupa i obejmuje pary od 6 do 12 chromosomów; Chromosomy X również należą do tej grupy. Większość z nich jest średniej wielkości i sub-metacentryczna. Wzorce paskowania pomagają w identyfikacji poszczególnych chromosomów.
(4) Grupa D:
Do tej grupy należy 13-15 par chromosomów. Wszystkie są średniej wielkości i akrocentryczne. Ciało satelitarne jest przymocowane do wolnego końca krótkiego ramienia każdego chromosomu.
(5) Grupa E:
Obejmuje liczbę chromosomów od 16 do 18. Są to dość krótkie chromosomy sub-metacentryczne.
(6) Grupa F:
Do tej grupy należy 19 i 20 chromosomów w parach. Każda z nich jest krótka i metacentryczna.
(7) Grupa G:
Zawiera 21 i 22 pary chromosomów; Chromosom Y należy do tej grupy. Każda z nich jest bardzo krótka i akrocentryczna, a 21 i 22 chromosomy przedstawiają ciała satelitarne na krótkich ramionach. Odległe końce długich ramion chromosomu Y zawierają ciałka fluorescencyjne po barwieniu barwnikiem flurochromowym.
Punkty obserwacji:
(a) 1 do 3 chromosomów grupy A i 19, 20 chromosomów grupy F są metacentryczne.
(b) 13 do 15 chromosomów grupy D oraz 21, 22 i chromosomów Y z grupy G są akrocentryczne. Pięć par chromosomów obejmujących 13, 14, 15, 21, 22 zawiera ciała satelitarne; stąd zwane sat-ch.ro- mosome. Chromosomy Sat zajmują się organizacją jąderek.
(c) Reszta chromosomów jest sub-metacentryczna.
Lokalizacja genów na chromosomach:
Lokalizację genu na poszczególnych chromosomach ludzkich, choć trudne do określenia, można ocenić na podstawie analizy rodowodu, badając pacjentów z delecją chromosomu i badając segregację genów "markerowych" w rodzinach z określonym zaburzeniem dziedzicznym. Geny markerowe są częste w populacji ogólnej. Autosomalne cechy markera obejmują grupy krwi i niektóre białka surowicy.
Cechy markera sprzężonego z X obejmują ślepotę barw, grupę krwi Xg i w niektórych przypadkach niedobór dehydrogenazy glukozo-6-fosforanowej. Badania rodowodu wykazały ścisłe powiązanie między loci genów grupy krwi ABO i zespołem rzepki paznokci oraz między grupą krwi Duffy a jedną postacią wrodzonej zaćmy.
Mapowanie genów na poszczególnych chromosomach jest dalej ulepszane przez zastosowanie enzymów restrykcyjnych (endonukleazę), które są syntetyzowane przez wiele bakterii. Enzymy restrykcyjne dzielą DNA na fragmenty o zmiennej długości, przecinając specyficzną sekwencję zasad, których miejsca są różne dla różnych enzymów. Taki polimorfizm długości fragmentów restrykcyjnych (RFLP) działa jak odcisk palca DNA i jest wykrywany przez przyjęcie technologii rekombinacji DNA.
Analiza struktury DNA za pomocą RFLP umożliwia określenie, który z rodziców jest źródłem wadliwego chromosomu. Pomaga to w poradnictwie genetycznym, w badaniu przestępstw i ustalaniu ojcostwa. Ocenia się, że około 50 000-100 000 genów występuje w całym ludzkim genomie z 3 miliardami par zasad. Na początku 1993 r. Ponad 2500 loci przypisano do określonych pozycji na ludzkiej mapie genetycznej.
Nieprawidłowości około 450 tych genów zostały powiązane z ludzkimi chorobami. Część ważnej lokalizacji genów na autosomach znajduje się tutaj.
Chromosom:
1 - grupa krwi Dufy'ego, czynnik Rh, białka histonowe, wrodzone catract, barwnikowe zapalenie siatkówki.
2 - fosfataza kwaśna z krwinkami czerwonymi. Lekki łańcuch immunoglobuliny Kappa.
5 - Heksosaminidaza-B
6 - Główny kompleks zgodności tkankowej (HLA), ataksja spino-móżdżka, zespół adrenogenitalny.
7 - Gen strukturalny kolagenu.
9 - Grupa krwi ABO, zespół rzepki paznokci.
14 - Ciężki łańcuch immunoglobuliny
15 - Kinaza heksozoaminidazy-A 17-tymidyny
19 - Czułość wirusa polio i echa
20 - Deaminaza adenozyny
21 - gen zespołu Downa; gen dla choroby Alzheimera;
22 - Geny dla łańcucha lekkiego immunoglobuliny lambda
Wydaje się, że chromosom X zawiera loci dla dehydrogenazy glukozo-6-fosforanowej, hemofilii A, widzenia barwnego i dystrofii mięśniowej Beckera na długim ramieniu, oraz grupy krwi Xg, rybiej łuski, albinizmu ocznego i umiejscowionych w locie umiejscowionych umiejscowieniach umysłowych krótkie ramię.
Chromosom Y zawiera samce determinujące geny "SRY", składnik TDF (czynnik determinujący jądra). Obecność pojedynczego chromosomu Y indukuje rozwój jąder; testy płodu uwalniają testosteron i czynnik regresji mullerowskiej, które poprzez lokalne działanie pozwalają na różnicowanie kanalików mezenelitarnych i kanałów, aby rozwinęły się w system przewodów jąder i jednocześnie pomagają w regresji przewodów parameonephric (układ mullerowski). Zatem chromosom Y przez pociąg zdarzeń indukuje rozwój męskich gonad, płciowych przewodów i zewnętrznych genitaliów wyrażających męski fenotyp.
Ale w zespole "feminizacji jąder" z chromosomami XY osobnik wydaje się być doskonałą kobietą z piersiami i żeńskimi zewnętrznymi genitaliami, ale z jądrami w jamie brzusznej. Z powodu defektu genetycznego chromosomu Y, układ mullerowski przestaje reagować na działanie męskich hormonów uwalnianych przez jądra płodu.
Normalny samiec przedstawia budowę chromosomu XY; ale gdy dana osoba ma więcej niż jeden chromosom X z pojedynczym chromosomem Y (47, XXY; 48 XXXY), osobnik jest fenotypowo męski z dysgenezą kanalików nasiennych (zespół Klinefeltera). Dlatego chromosom Y przedstawia silne geny determinujące męskie, niezależnie od liczby chromosomów X. Jednak obecność dodatkowych chromosomów X w zespole Klinefeltera powoduje zmniejszenie płodności i sprawia, że osoba jest nieco upośledzona umysłowo.
Oprócz genów determinujących samce, chromosom Y zawiera geny dla owłosionej pinny i antygenu HY (zgodności tkankowej). Długość chromosomu Y różni się w zależności od osoby i jest zgodna z zasadą Mendelizmu. Z powodu obecności antygenu HY, przeszczepy męskie są czasami odrzucane przez samice tego samego szczepu.
Normalna samica posiada konstytucję chromosomu XX. We wczesnej embriogenezie oba chromosomy X są aktywne genetycznie i indukują rozwój jajników. Następnie jeden chromosom X staje się heterochromatyczny i genetycznie obojętny i utrzymuje się jako chromatyna płciowa lub ciało Barr (heterochromatyna o działaniu heterogenicznym). Jajniki płodowe nie wydzielają żadnego hormonu. Dlatego przy braku jąder (z lub bez jajników) system wolffia (mezonephric) cofa się, a system Mullerian (parameonephric) różnicuje się w żeńskie narządy płciowe i żeńskie zewnętrzne narządy płciowe.
W rzadkich przypadkach osoba z konstytucją chromosomu XX wydaje się być mężczyzną o fenotypie; to sugeruje obecność genów determinujących jądra w jednym z dwóch chromosomów X, które pochodzą od Y. To rzadkie dziedziczenie jest możliwe u osobnika z powodu krzyżowania się w gametogenezie po stronie ojcowskiej. Zaskakująco zaobserwowano, że osoby z konstytucją chromosomu 45, XO mogą pozostać przy życiu, ale połączenie 45, YO nie jest opłacalne.
Strukturalna zmiana chromosomów (ryc. 11-12):
Usunięcie:
Oznacza to utratę odcinka chromosomu, który może być końcowy lub śródmiąższowy. Usunięcie interstitalu wynikające z dwóch przerw prowadzi do zerwania zerwanych końców. W zespole "cri du chat" kasowana jest część końcowa krótkiego ramienia chromosomu 5.
Translokacja:
Wymiana segmentów między chromosomami nie homologicznymi jest znana jako translokacja. Proces translokacji wymaga przerw w obu niehomologicznych chromosomach, po których następuje naprawa prowadząca do nieprawidłowego układu. Translokacja nie zawsze może wywoływać nieprawidłowy fenotyp, ale może prowadzić do powstawania niezrównoważonych gamet i niesie wysokie ryzyko nieprawidłowego potomstwa.

Wzajemna translokacja pomiędzy dwiema parami niehomologicznych chromosomów może być heterozygotyczna, gdy bierze się tylko jeden z chromosomów w parze, lub homozygotyczna, gdy obydwaj członkowie pary chromosomów wymieniali między sobą segmenty. Czasami translokacja obejmuje trzy przerwy, a uszkodzona część chromosomu jest wprowadzana do niehomologicznych chromosomów, podczas gdy inny niehomologiczny chromosom przedstawia śródmiąższową delecję.
Translokacja Robertsona lub fuzja centryczna jest specjalnym rodzajem translokacji, w której dochodzi do zerwania w centromerach dwóch chromosomów i wymienia się całe ramiona chromosomu. U mężczyzny zwykle obejmuje on dwa akrocentryczne chromosomy, np. Między grupami D i G, 21/22 lub 21/21. W translokacji D / G długie ramię chromosomu G jest połączone z długim ramieniem chromosomu D, a fragment utworzony przez połączenie krótkich ramion dwóch chromosomów zostaje utracony.
Matka z translokowanym zespołem Downa jest zwykle nosicielem translokacji D / G tylko 45 chromosomami. Wytwarza cztery rodzaje gamet - jedną z normalnym chromosomem D, jedną ze zwykłym chromosomem G, jedną z translokowanym chromosomem D / G podobnym do matki nośnej i jedną z chromosomem D / G i normalnym chromosomem G.
Potomstwo pochodzące z ostatniej odmiany gamet będzie mieć 46 chromosomów, ale będzie trisomiczne dla chromosomu 21 z manifestacją zespołu Downa. Dlatego matka-matka z translokacją D / G będzie miała ryzyko urodzenia dziecka z zespołem Downa. Kiedy matka niesie translokację obejmującą oba chromosomy 21, wszystkie jej dzieci będą miały zespół Downa.
Odwrócenie:
Część chromosomu zostaje odłączona, a następnie łączy się z tym samym chromosomem w pozycji odwróconej. Geny nie są tracone, ale umieszczane w zmienionych loci.
Izo-chromosom:
Centromer chromosomu, z powodu nieprawidłowej anafazy (mitozy lub mejozy), dzieli się poprzecznie zamiast podłużnego podziału. Kulminuje to powstanie dwóch chromosomów o różnej długości, z których każda przedstawia chromosomy metacentryczne z duplikacją genów. Otrzymane chromosomy pochodzące z poprzecznego podziału centromeru są znane jako izochromo- somy.
Powielanie:
Jest to proces dodawania części chromosomu z innego chromosomu homologicznego z duplikacją genów. Efekty związane z duplikacją genów związane z izosplitowaniem chromosomów X są czasem obserwowane w zespole Turnera.
Chromosom pierścienia:
Chromosom pierścieniowy jest obserwowany, gdy chromosom jest usuwany na obu końcach, a następnie usunięte "lepkie" końce są przylegające do siebie w formie pierścienia. Manifestacja chromosomu pierścieniowego zależy od delecji określonych genów.
Symbole używane w Cytogenetic:
p-Krótkie ramię chromosomu
q-długie ramię chromosomu
t-Translokacja; inv-Inversion
i-Iso-chromosom;
chromosom r-pierścienia
+ lub -Sign: Po umieszczeniu przed odpowiednim symbolem, oznacza to dodanie lub brak całego chromosomu. Na przykład zespół trisomii 21 Downa może być reprezentowany jako 47, XY + 21.
Kiedy + lub - singns są umieszczane za symbolem, oznaczają wzrost lub spadek długości chromosomu. Na przykład syndrom Cri-du-chat wpływający na dziecko płci męskiej z delecją krótkiego ramienia chromosomu 5 jest przedstawiony jako 46, XY, 5p
W filadelfii lub chromosomie Ph 'występuje translokacja wzajemna między długim ramieniem chromosomu 9 w paśmie 34 i długim ramieniem chromosomu 22 w paśmie 11. Dlatego kariotypem tej choroby jest -t (9; 22) (q34; ql 1).
Notacja jest dodatkowo dopracowywana w celu wskazania konkretnych pasm na dowolnym określonym chromosomie.
Linia przekątna na chromosomach lub ich liczba wskazuje na mozaikę, np. XY / XX; XO / XX; XY / XXX; 45/46/47.
Geny:
Geny są jednostkami dziedziczności i składają się z części określonych cząsteczek DNA. Jak wspomniano wcześniej, geny są ułożone w szeregi liniowe w chromosomach z dokładną sekwencją i liczbą podstaw DNA, różne dla różnych genów i mające zdefiniowany początek i zdefiniowane zakończenie. Ponieważ pojedynczy chromosom zawiera jedną podwójną helisę cząsteczki DNA w ciasno zwiniętej postaci, liczne geny lub cistrony są przenoszone przez pojedynczą cząsteczkę DNA.
Pozycja genu w chromosomie nazywa się locus, co mierzy się w odniesieniu do centromeru. Zwykle geny nie zmieniają loci, z wyjątkiem rekombinacji podczas krzyżowania lub zmiany morfologii chromosomów.
Geny zajmujące identyczne loci w parze homologicznych chromosomów nazywa się allelomorfami lub allelami. Ogólnie rzecz biorąc, geny alleliczne regulują różne cechy fizyczne i biochemiczne jednostki. Z punktu widzenia molekularnego jedna para genów allelicznych reguluje syntezę jednego łańcucha polipeptydowego.
Kiedy geny alleliczne regulujące konkretną postać lub cechę, np. Wzrost, działają w tym samym kierunku (zarówno wysokie, jak i krótkie), nazywane są homozygotami; podczas pracy w przeciwnym kierunku (jeden wysoki, a drugi krótki) allele są heterozygotyczne. Większość dziedzicznych cech jest poligenicznych i jest wytwarzana przez złożone oddziaływanie wielu genów i wpływ środowiska. Czasami para genów allelicznych może wpływać na więcej niż jedną postać; jest to znane jako plejotropia.
Struktura chemiczna DNA (ryc. 11-13):
Zostało ustalone w 1953 roku przez Wilkinsa, Watsona i Cricka na dyfrakcji rentgenowskiej, że cząsteczka DNA składa się z dwóch pasm polinukleotydów ułożonych w podwójną helisę. Każda nić składa się ze szkieletu altenowego cukru pentozowego (D-2-dezoksyryboza) i cząsteczki fosforanu, a dwie nici są utrzymywane razem przez wiązania wodorowe między podstawami azotowymi, które są przyłączone do cukrów jako grupa boczna i skierowane w stronę centrum spirali.
Podstawy są dwóch rodzajów, puryn i pirymidyn. Puryna w jednej nici zawsze łączy się z pirymidyną w drugiej nici. Zasady purynowe obejmują adeninę (A) i guaninę (G); zasady pirymidynowe obejmują tyminę (T) i cytozynę (C). Parowanie zasad jest specyficzne w normalnych warunkach (gdy występuje w postaci keto) - pary paradynowe z tyminą mającą dwa wiązania wodorowe i jest reprezentowane przez A = T; pary guaniny z cytozyną za pomocą trzech wiązań wodorowych i reprezentowane przez G = C.
Pokazuje to, że podczas de- naturacji DNA rozdział dwóch nici na poziomie A = T jest szybszy niż poziom G = C. Jednakże, gdy zasady są w postaci enolowej, adenina może łączyć się z cytozyną i guaniną z tyminą. Jest to podstawą mutacji genów.

Dwie nici cząsteczki DNA są komplementarne względem siebie. Jeśli znana jest sekwencja zasad jednej nici, można sformułować skład podstawowy drugiej nici. Sekwencja zasad i liczba nukleotydów DNA są specyficzne i różnią się w różnych genach. Tak więc istnieją niezliczone formy DNA w genach i przechowują różnorodne informacje genetyczne.
Funkcje cząsteczki DNA:
Cząsteczki DNA mają następujące możliwości:
(1) Self Replication
(2) Biosynteza RNA i białek
(3) Rekombinacja;
(4) Mutacja.
Samodzielna replikacja (ryc. 11-14):
Podczas podziału jądrowego dwie nici cząsteczki DNA oddzielają się, a każda nić działa jako matryca i organizuje tworzenie nowej komplementarnej nici z puli nukleotydów w wyniku specyficznego parowania zasad. W ten sposób, gdy komórki dzielą się, informacje genetyczne są przesyłane w niezmienionej postaci do każdej komórki potomnej. Obie nici uczestniczą w procesie replikacji DNA, który odbywa się w fazie S (syntezie) cyklu komórkowego. Replikacja obejmuje kilka enzymów, takich jak polimeraza DNA, ligaza DNA i specyficzna endonukleaza.

Biosynteza RNA i białek:
Cząsteczka DNA działa również jako matryca do syntezy RNA, a ta ostatnia przekazuje informację genetyczną i odszyfrowuje syntezę specyficznego łańcucha polipeptydowego białek przez liniowe łączenie aminokwasów. Dlatego centralny dogmat genetyki molekularnej obejmuje DNA → RNA w procesie transkrypcji oraz RNA → białka w wyniku translacji.
RNA (kwas nukleinowy rybozy) różni się od DNA zasadniczo trzema sposobami: posiada zwykle jednoniciowy łańcuch polinukleotydowy; cukier pentozowy to D-ryboza; spośród czterech zasad organicznych trzy są podobne do DNA (Adenina, Guanina, Cytozyna), a czwarty to uracylo zamiast tyaminy. Dlatego podczas transkrypcji z DNA do RNA pary adeninowe z urydylem (A = U). RNA występuje w trzech postaciach - informacyjnym RNA (mRNA), rybosomalnym RNA (rRNA) i transferowym RNA (tRNA). Polygenic cząsteczka DNA działa jako szablon dla wszystkich trzech odmian RNA. W przeciwieństwie do replikacji DNA, tylko jedna z dwóch nici cząsteczki DNA działa jako matryca dla RNA.
Łańcuch polinukleotydowy mRNA jest tworzony w jądrze przez jedną dowolną nić cząsteczki DNA przy pomocy polimerazy RNA. Podczas syntezy RNA dwie nici DNA oddzielają się (ryc. 11-15). Selekcja nici DNA do syntezy RNA odbywa się za pomocą polimerazy I RNA dla rRNA, polimerazy II dla mRNA i polimerazy III dla tRNA. Tak utworzony RNA komunikacyjny przekazuje informację genetyczną o komplementarnej sekwencji zasad i przenosi się do cytoplazmy przez pory jądrowe.

Szereg cytoplazmatycznych rybosomów (zawierających rybosomalne RNA i białka) jest związanych z łańcuchem polinukleotydowym mRNA. Rybosomy są miejscami, w których łańcuchy polipeptydowe białek są tworzone przez liniowe wiązanie różnych aminokwasów.
Sekwencja aminokwasowa i liczba są specyficzne dla różnych białek; są one określone przez dokładny odczyt podstawowej sekwencji mRNA w kierunku końca 5 'do 3'. W biosyntezie białek bierze udział dwadzieścia (20) aminokwasów. Przed utworzeniem wiązania peptydowego aminokwasy są aktywowane i przyłączone do jednego końca specyficznej cząsteczki przenoszącego RNA (tRNA). Podstawowa sekwencja aktywowanych aminokwasów tRNA identyfikuje komplementarną sekwencję zasad mRNA i jest przyłączona do tej ostatniej przez wiązania wodorowe aż do utworzenia łańcucha polipeptydowego białka.
Dlatego mRNA, rRNA, tRNA i wiele enzymów aktywnie uczestniczy w różnych etapach biosyntezy białka. Skomplikowany proces biosyntezy z łańcucha polinukleotydowego mRNA do łańcucha polipeptydowego białka jest znany jako translacja (Fig. 11-16). Łańcuch polinukleotydowy mRNA może być monokloniczny lub policistronowy.

Kody genetyczne:
Ponieważ zasady DNA lub RNA i aminokwasów białek są ułożone w liniową sekwencję, musi istnieć pewien związek między azotowymi zasadami i aminokwasami. DNA lub RNA przedstawia cztery (4) zasady, a pierwotna struktura białek składa się z dwudziestu (20) aminokwasów. Po pracochłonnych eksperymentach Nirenberg i Matthaei w 1961 roku ustalili, że sekwencja trzech (3) zasad mRNA (a więc i komplementarnego DNA) koduje jeden aminokwas.
Ponieważ trzy kolejne zasady są specyficzne dla jednego aminokwasu, możliwa liczba kombinacji czterech zasad wziętych po trzy razy na raz wynosi 4 3 lub 64. Taki triplet oparty na nukleotydach jest nazywany kodonem. Ostatecznie odkryto wszystkie 64 kodony określające inny aminokwas. Jednakże trzy kodony, takie jak UAG, UGA i UAA, nie kodują żadnego aminokwasu; stąd te trzy są nazywane nonsensem lub terminalnymi kodonami i sygnalizują zakończenie łańcucha polipeptydowego.
Znane są trzy niesparowane zasady przyłączone do jednej pętli tRNA
jako anty-kodony, które pasują do komplementarnych kodonów mRNA. Mając na uwadze, że kodony są odczytywane od końca 5 'do końca 3', odczyty antykodonów są odczytywane z kierunku 3 'do 5'; jak wspomniano wcześniej, tRNA przenosi aktywowany aminokwas na jednym końcu łańcucha.

Kod genetyczny mRNA i aminokwasy, dla których kodują.
Kodon jest zgodny z niektórymi zasadami:
(a) Kodony nie nakładają się i zachowują ścisłą sekwencję wzdłuż nici polinukleotydowej mRNA.
(b) Są uniwersalne i mają zastosowanie do wszystkich organizmów.
(c) Kodony degeneracyjne - kiedy dwa lub więcej kodonów oznacza ten sam aminokwas, mówi się, że ma postać degeneracyjną. GUU, GUC, GUA, kod GUG dla Valine; UUU, kod UUC dla fenyloalaniny; UUA i UUG oznaczają leucynę. W większości przypadków dwie pierwsze zasady pozostają nienaruszone, a zmiana trzeciej bazy powoduje degenerację.
(d) Niejednoznaczny lub nieprawidłowy kodon określa różne aminokwasy. W normalnych warunkach UUU oznacza fenyloalaninę, ale w obecności streptomycyny może kodować leucynę lub izoleucynę.
(e) Rozpoczęcie lub uruchomienie kodów kodonu-AUG dla metioniny i działa jako sygnał startowy w syntezie łańcucha polipeptydowego. Sekwencja aminokwasów w łańcuchu polipeptydowym jest znana jako pierwotna struktura białka.
Wolna grupa aminowa na jednym końcu łańcucha jest znana jako koniec N, a wolna grupa karboksylowa na drugim końcu łańcucha jest nazywana końcem Ń. Każdy aminokwas w łańcuchu nazywany jest resztą. Reszta końca N jest uważana za pierwszą liczbę, a reszta na końcu C jako ostatnia liczba sekwencji aminokwasowej.
Metionina w kompleksie inicjacyjnym jest formylowana przez specyficzne enzymy tak, że wiązanie peptydowe nie zachodzi na N-końcu. Dwa kodony, AUG i UGG oznaczają tylko jeden aminokwas; AUG dla metioniny i UGG dla tryptofanu.
(f) Kodon końcowy lub niesekwencyjny. Trzy kodony, takie jak UAG, UGA i UAA, nie kodują żadnych aminokwasów. Terminalne kodony oznaczają zakończenie łańcucha polipeptydowego.
Aktualna koncepcja organizacji genów:
1. Jak wspomniano wcześniej, gen jest częścią specyficznej cząsteczki DNA, która reguluje syntezę jednego łańcucha polipeptydowego. Typowy gen składa się z nici DNA, która zawiera jednostkę transkrypcyjną i region promotora.
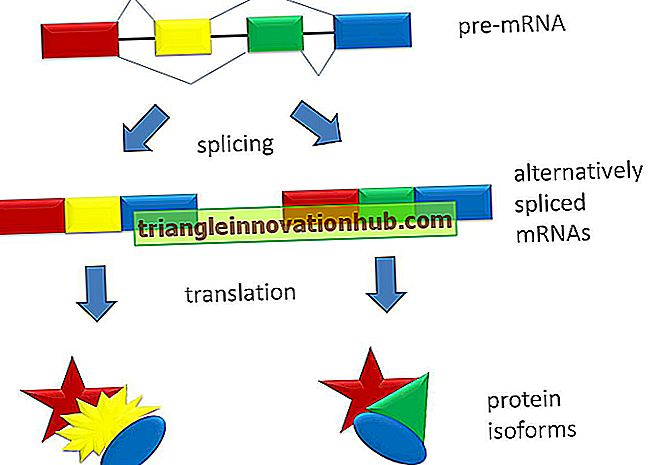
Jednostka transkrypcji składa się z kilku odcinków eksonów, które dyktują tworzenie białek, rozdzielonych przez segmenty intronów, które nie ulegają translacji na białka. Pre-mRNA powstaje z DNA, a następnie introny są eliminowane w jądrze przez proces potranskrypcyjnego splicingu, tak że końcowy mRNA, który wchodzi do cytoplazmy, składa się tylko z eksonów.
The promoter region lies on the 5′ end side of the transcription unit of the gene. It contains various DNA segments which precede the transcription unit from 3′ end to 5′ end side in the form of Specifier, Quantifier and Regulator segments. The base sequence of specified segment includes TATA (popularly called TATA Box), which ensures that the transcription starts a proper point. Z-DNA is a segment of promoter region, that may determine tissue-specific expression.
2. Post-translational modification. After polypeptide chain is translated through mRNA, rRNA and tRNA, the final protein product is modified by a combination of reactions that include hydroxylation, carboxylation, glycosylation or phosphorylation of amino acid residues. A larger polypeptide is converted to a smaller form by cleavage of peptide bonds; thereafter the protein is folded into its complex configuration.
A typical eukaryotic cell synthesizes about 10, 000 different proteins during its life time. Proteins synthesized by the genes may be one of three types—enzymes, structural proteins and regulatory proteins.
3. The cytogenetic analyses in hydatidiform mole, a tumour or trophoblastic, suggest that the abnormal ovum loses its own nucleus and is fertilised by two sperms. Thus the zygote contains two male pronuclei, possessing between them at least one X chromosome, In complete molar pregnancy trophoblastic membranes develop, but embryos do not appear, Genomic imprinting suggests that the maternal chromosomes regulate embryoblast development, and the paternal chromosomes regulate trophoblastic development.
Recombination:
During crossing over in meiosis, there is exchange of genetic material between homologous chromosomes. This leads to recombination or shuffling of genes. One of the two events might be observed in crossing over. The two different genes that were originally located on the same chromosome of a particular chromosome pair, might be separated from each other and thereafter are distributed to both homolgous chromosomes; or one of the two genes originally located in each homologous chromosome might be brought together on the same chromosome.
When two different genes are located on the same chromosome pair, they are said to be linked. Crossing over is more likely to occur between the genes on a particular chromosome which are far apart than the genes which are close together. One can assess the relatives distances between genes on any chromosome by determining the frequency with which crossing over takes place between these genes. Genetic distance between two loci on a particular chromosome is expressed in centimorgan (cM). Two loci are 1cM apart, if there is a 1% probability of cross over between them in meiosis. On an average 30 to 35 cross over per cell are estimated to occur during meiosis in males, and perhaps twice as many during meiosis in females.
By determining the frequency of recombination due to cross-over among the progeny, it is possible to frame a linkage map in human with the grouping of genes on particular chromosomes. (Vide supra, in gene localisation on chromosomes)
Recombination of DNA fragments can be studied experimentally by allowing fusion of cells from two different species and then placing in culture. The fused cell-hybrids contain chromosomal constitution from both species and exchange segments of DNA as they regenerate and divide. All these regeneration processes involve random exchange of DNA sequences, and eventually protein synthesis is significantly changed from the pre-fused ancestor cells.
In 1972, Jackson et al. described the biochemical methods for cutting DNA molecules from two different organisms, using restriction enzymes, and recombining the fragments to produce biologically functional hybrid DNA molecules.
Subsequently, scientists successfully inserted the genes for both chains of insulin into some strain of Escherichia Coli, and after isolation and purification, the A and В chains were joined by disulphide bonds to produce human insulin. With the discovery of 'Recombinant DNA' technology, a number of essential substances such as, human insulin, interferon, human growth hormone, calcitonin and many others are produced commercially.
Mutation:
A change of a base pair of the DNA molecule is known as the gene mutation (point mutation). Since genes are responsible for the synthesis of protein through transcription from DNA to RNA and translation from RNA to protein, the mutation may have the following diverse effects on the corresponding protein:
(a) The changed triplet codon may code for the same amino acid without any alteration of the resulting protein. About 20 to 25% of all possible single base changes belong to this type.
(b) In about 70 to 75% cases a single base mutation may code for a different amino acid and result in the synthesis of an altered protein which produces reduced or complete loss of biological activity.
(c) In about 2 to 4% cases of single base mutation, the triplet may signal the termination of a peptide chain which is unable to retain normal biological activity.
(d) On rare occasions, more than a single base in the DNA sequence may be involved in a gene mutation. As a result the level of a particular enzyme may be reduced because it is not synthesized or synthesized with reduced activity. Sometimes, a gene mutation may lead to increased synthesis of enzymes with increased activity.
(e) In some cases of genetic disorders a specific protein may be synthesized, but the protein remains functionally inactive. This happens in most cases of haemophillia.
Normally base pairing in replication or transcription takes place in keto form, where the combinations are A=T (in DNA), A=U (in RNA), G=C. But in a gene mutation base pairing occurs in enolfrom, in which combinations are A=C, G=T (in DNA), G=U (in RNA). Such unusual bair pairing is known as tautomerisation.
Mutation may be spontaneous or induced by various chemical or physical agents, eg mustard gas, radiation from X-rays, gamma rays from radium and other radio-active atoms. Mutant genes may be inherited or appear at random. One of the typical examples of a gene mutation is observed in sickle-cell anaemia, where the beta chain of adult haemoglobin containing 146 amino acids possesses Valine, instead of glutamic acid, in the 6th position.
RNA directed DNA synthesis:
It has been suggested by Temin in 1972, from the study of RNA viruses that the flow of genetic information occasionally occurs in reverse direction from RNA to DNA with the help of reverse transcriptase. Such viruses are known as retroviruses, which when introduced into the host animal cell incorporate with the specific region of strand of nuclear DNA by a process of recombination.
This forms the basis of the study of oncogenes. Certain regions of DNA in normal cells serve as templates for the synthesis of RNA and the latter in turn acts as template for the synthesis of DNA, which is subsequently incorporated with the nuclear DNA. The resultant amplification of certain regions of DNA helps in embryonic differentiation and possibly in the pathogenesis of cancer.
Types of Genes:
1. Dominant gene expresses its physical or biochemical trait, when the allelic genes are either homozygous or heterozygous for the trait. This follows Men- delian patterns of inheritance and can be observed from the pedigree record of the family. Tallness is caused by dominant gene. The genetic constitution of a tall individual may be T:T or T:t (T for tallness, t for shortness). Most of the dominant traits are expressed in heterozygote state (Fig. 11-17).

Genetic disorders caused by mutation of autosomal dominant genes possess the following characteristics:
(a) The trait is passed on from one generation to another. It has a vertical transmission. Each affected person usually has an affected parent. Sometimes the disorder may appear suddenly in one generation. This may result from a fresh mutation; or if the parent with abnormal gene died in early life before the disease could manifest, the history of parent's affection may be lacking. This is so in Huntington's chorea, where the disease is expressed in mid-adult life.
(b) When one of the parents is affected, the risk of having an affected child is 50%.
(c) Since the trait is autosomal, both sexes may be equally affected. Some autosomal genes are expressed preferentially in one sex. These are called sex- limited genes. Gout and pre-senile baldness predominantly affect the males.
(d) If the affected individual marries a normal person, half of their children will be affected.
(e) The degree of expression of abnormal trait may vary in different members of the same family. For example, in poly- dactyly some member shows a small wart-like appendage on the side of the hand, whereas other member exhibits a complete extra-finger. Sometimes a gene, when non-penetrant, may not express it at all. If a child and a grandparent have the same disease and the middle generation does not show any manifestation, the condition is said to have skipped a generation.
(f) The unaffected mambers of a family do not transmit the trait further.
2. Co-dominant genes:
When both the allelic genes are dominant but of two different types, both traits may have concurrent expression. In ABO blood groups, A gene and В gene are both dominant; when they occupy identical loci in homologus chromosomes, AB blood group is expressed (Fig. 11-18).

3. Recessive genes:
Expresses the trait only in homozygote state that means when both alleles are recessive for that trait (Fig. 11- 19). Therefore following the Mendelian principles the genetic constitution of a short individual is t:t (t for shortness).

Diseases caused by mutation of autosomal recessive genes present the following characteristics:
(a) The disease is transmitted by a couple both of whom are carriers of one abnormal gene, but are themselves healthy because the other allele is normal.
(b) The pattern of transmission appears horizontal in pedigree analysis because often siblings are affected, whereas the parents are normal.
(c) The risk of having an affected child (with the double dose of the abnormal gene) to a carrier couple is 25%. Therefore, most carrier couples, if advised properly, would not take the risk of having another affected baby unless prenatal diagnostic facilities are available for the trait.
(d) Most of the metabolic abnormalities are inherited as autosomal recessive traits. The heterozygote status of a carrier couple (having one affected child) may be detected biochemically in several inborn errors of metabolism. The enzyme levels in the heterozygotes are about 50% less than the control.
(e) Since the condition is autosomal, both sexes are likely to be affected equally.
(f) The parents of individuals affected with autosomal recessive traits are often related, since the marriages between close blood relatives (cousin marriages) are more likely to carry the same genes from a common ancestor. The rarer a recessive disease, greater is the frequency of consanguinity among the parents of affected individuals.
(g) If two persons homozygous for a recessive condition were to marry and have children, all their children would be affected. But this is not so in every case. In one family both parents were albinos (recessive disorder), but their children were normal; careful examination of the father revealed that he had a different type of albinism from his wife.
4. Carrier Gene:
Heterozygous recessive gene acts as a carrier which may be expressed in subsequent generations. When both parents are heterozygous tall (T:t), the possibilities of the height of the offspring may be such that out of four children three are tall and one short, in the proportion of 3:1. One tall child is homozygous, and the other two are heterozygous.
5. Sex-linked Genes:
The genes located on the X chromosome or Y chromosome are known as the sex- linked genes. Mutation of X-linked genes is more common, and is mostly expressed as recessive traits.
Х-linked recessive traits (Fig. 11-20):
Haemophilia, partial colour blindness, glucose- 6-phosphate dehydrogenase deficiency, Duchenne's muscular dystrophy are examples of X-linked mutant recessive genes. These traits exhibit the following characteristics:

(a) The female (XX) becomes carrier of the disease when one X chromosome contains an abnormal gene, whereas the allelic gene of other X chromosome is normal. So the females do not express the disease in heterozygous state. On the other hand, when the abnormal gene involves the non-homologous part of single X chromosome of a male (XY), the disease is expressed in that individual because the defective gene has no corresponding allele in Y chromosome to counter-act. Hence, the affected male is called hemizygous. Broadly speaking, in X linked recessive traits the females are the carriers and the males are the victims of the disease.
(b) When mother is a carrier and father is healthy, 50% of the sons are affected by the disease and the remaining 50% are normal; 50% of the daughters are carrier of the disease and the rest are free. Therefore, when a boy is haemophilic, his mother should be a carrier and 50% of his sisters are carriers of the disease. However, the healthy brother or carrier-free sister of a haemophilic individual does not transmit the disease to the next generation.
(c) When mother is a carrier and father is haemophilic, half of the sons are affected and half are healthy; half of the daughters are affected and half are carriers. This shows that females may be affected in such parental combination, but the possibility is remote because the haemophilic male usually dies early before attaining parenthood. The above combination further shows that there is no male to male transmission.
(d) If the affected males do not reproduce, the pedigree pattern of an X-linked recessive trait tends to be oblique because the trait is transmitted to the sons of carrier sisters of affected males.
(e) On rare occasions, a female may exhibit X-linked recessive trait. This can be explained as follows:
(i) She might be a Turner female (XO);
(ii) Physically female appearance is due to testicular feminisation with XY chromosomes;
(iii) Affected female may have carrier mother and affected father; or a carrier mother and a normal father with fresh mutation affecting X chromosome.
X-linked Dominant Traits:
These are observed in Vitamin D resistant rickets and Xg blood group. The characteristics of dominant traits are as follows:-
(a) An affected male transmits the disease to all his daughters, but to none of his sons.
(b) Both males and females are affected, but the disease is less severe in females.
Y-Iinked Inheritance:
This is also known as the holandric inheritance, where only males are affected. The affected male transmits the trait to all his sons, and to none of his daughters. Male to male transmission is suggestive of Y-linked inheritance.
Hairy pinna and HY histocompatibility antigen manifest holandric inheritance.
Symbols used in Pedigree Chart (Fig. 11-21):

Autosomal Dominant Inheritance (Fig. 11- 22)

Some examples of autosomal dominant traits—
ja. Achondroplasia;
ii. Osteogenesis imperfecta;
iii. Brachydactyly, polydactyly, syndactyly;
iv. True pophyria with port-wine urine due to porphyrins;
v. Sex limited, gout and baldness affecting predominantly pre-senile males;
vi. Huntington's chorea, appearing at about 50 years or after;
vii. Angioneurotic oedema;
viii. Familial hypercholesterolaemia;
IX. Diabetes insipidus;
x. Marfan's syndrome, manifested by elongated extremities, dislocation of lens of eyes and cardio-vascular abnormalities;
xi. Nail patella syndrome, manifested by dystrophy of nails, absence of patella and nephropathy;
XII. Multiple neurofibromatosis;
XIII. Polyposis coil.
Some examples of X-linked recessive traits-
ja. Haemophilita-This is due to functionally defective antihaemophilic globulin.
ii. Partial colour blindness-It is expressed as inability to distinguish between red and green.
iii. Duchenne's muscular dystrophy.
iv. Glucose-6-phosphate dehydrogenase deficiency-It is manifested by haemo- lytic anaemia when treated with primaquin, phenacetin, nitrofurantoin, some sulphonamides and acetyl salicylic acid.
v. Testicular feminisation.
vi. Hunter's syndrome-It is due to deficient enzyme induronosulphate sulphatase, and is manifested by feature of Hurler's syndrome except corneal clouding.
Autosomal Recessive Inheritance (Fig. 11- 22) 23):

Some Examples of autosomal recessive traits—
(1) Inborn errors of metabolism;
ja. Acatalasia, due to deficient enzyme catalase; it leads to oral sepsis;
ii. Albinism, a complete depigmentation of ' skin due to deficiency of tyrosinase;
iii. Alkaptonuria, in which affected persons excrete dark coloured urine due to the presence of homogentisic acid. It is caused by the lack of enzyme homogentisic acid oxidase;
iv. Galactosaemia, due to deficiency of Galactose-I-phosphate uridyl transferase, and is manifested by vomiting and diarrhoea as a result of intolerance to galactose; this is followed by mental retardation, cataracts and cirrhosis of liver;
v. Hurler's syndrome, caused by deficient enzyme iduronidase, and manifested by mental retardation, skeletal abnormalities, hepatosplenomegaly and corneal clouding.
vi. Phenylketonuria, due to lack of phenylalanine hydroxylase and manifested by mental retardation, fairy skin and epilepsy;
vii. Tay-sachs disease, due to lack of hexosaminidase, and manifested by mental retardation, blindness and neurological abnormalities.
(2) Haemoglobinopathies:
ja. In sickle-cell anemia, beta chain contains valine in the 6th position, instead of glutamic acid. Heterozygous sickle cell-traits are more resistant to the attacks of malaria.
ii. Thalassemia major is expressed in homozygotes, and thalassemia minor in heterozygotes.
(3) Immunoglobinopathies:
ja. Some of the immunological disorders may be due to autosomal recessive traits.
X-linked Recessive Inheritance (Fig. 11-24):

Genetic Factors in Some Common Diseases:
Cukrzyca:
Early onset diabetes (juvenile-IDDM) is more genetically predisposed than late onset diabetes. Some investigators suggest that is possesses autosomal recessive inheritance, while others believe that is has multifactorial inheritance. In genetically predisposed individuals, prediabetics are recognised by raised serum level of islet-cell antibodies.
Nadciśnienie tętnicze:
There are two schools on the modes of inheritance; one school suggests that it possesses multifactorial inheritance, whereas other school believes that it is due to mutation of single dominant gene.
Ischaemic heart diseases:
Early onset ischaemic heart disease is due to familial hyper- cholesterolaemia which is inherited as an autosomal dominant trait. In the majority of the affected individuals the condition is multifactorial with a heritability of about 65%.
Peptic ulcer:
Duodenal ulcer is more frequent in individuals with О group of blood and in non-secretors of ABO substance. Forty per cent of the peptic ulcers possesses hereditary predisposition.
Schizophrenia:
It is inherited on a multifatorial basis with a heritability of about 85%. Some believes that it is inherited as autosomal dominant traits.
Some Terminology used in Genetics
(1) Genome:
The genome indicates the full set of genes, haploid in the gametes and diploid in the somatic cells of an individual.
(2) Genotype:
It means the genetic constitution of an individual which is fixed at the time of fertilization. The genotype of a tall individual may be T:T (homozygous) or T:t (heterozygous) which may be assessed by pedigree analysis.
(3) Phenotype:
It means physical or biochemical expression of the genotype. Phenotype is potentially variable and is the result of interaction between the genotype and the environment in which the individual develops and grows. It may so happen that an individual with T:T genotype is short in height. This is presumably due to some endocrinal or nutritional disorders which suppress the action of genotype.
(4) Phenocopy:
Sometimes a change in the environment produces a new phenotype, which closely resembles the appearance causes by a specific genotype. Such form of phenotype is known as phenocopy.
Jumping Genes or Transposons:
These are groups of genetic elements that really can move from place to place and by doing so modify or suppress the function of target genetic region. The jumping genes include pseudogenes, the retroviruses and oncogenes, and possess DNA sequences that jump. Each jumping gene has a short, similar, terminal repeat of bases at either end.
Each has the property of recognition of a specific sequence on the target DNA and generates a direct repeat of the same. Coming to the target sequence, the movable genes produce asymmetrical breaks on the opposite strands of the DNA duplex and then are integrated in the target site.
The transposons control mutation and recombination, and may be responsible for amplification of genes. The functional status of the jumping genes is still inconclusive.
Genetic Counselling:
Whenever an individual or a couple with genetic disorder seeks advice, the genetic counsellor is confronted with three problems;
(a) To establish the precise diagnosis (genetical or environmental) by clinical examination and laboratory investigations;
(b) To discuss the prognosis and value of any possible treatments;
(c) To determine the risk of recurrence of the disease in a family and to investigate the carrier detection, if any.
Chromosomal studies and karyotypes are indicated in the following conditions;
(i) In infants with congenital anomalies involving more than one system;
(ii) In abnormal sexual development;
(iii) Infertility, recurrent abortions etc.
When chromosomal defects (numerical or structural) are detected with abnormal phenotypes, treatment, if any, is symtomatic and not curative.
Discussion about the Risk of Recurrence in a Family:
(1) If both parents have normal chromosomes, although the child is affected by chromosomal abnormality (say, Trisomy 21-Mongol), the parents may be assured that the chances of recurrence of the same condition affecting future children are less, because the cause of this abnormality is non-disjunction in gametogenesis particularly involving elderly mother, and the phenomenon is mostly accidental.
If, however, the karyotype of Mongol baby shows translocation between G and D chromosomes (46) and the karyotype of the healthy mother shows balanced translocated chromosomes, the parents should be informed that similar Mongol baby might develop more frequently in subsequent pregnancies.
(2) In affected person with heterozygous autosomal dominant gene (say, Achondroplasia), the risk of recurrence among the offspring is 1 in 2 (50%), provided the dominant gene is fully penetrant.
(3) In autosomal recessive disorders, when both parents are healthy with heterozygous recessive gene for the same trait, chance of recurrence (say, phenylketonuria) among the offspring is 1 to 4. All of us are carrying about 3 to 8 detrimental recessive genes, but chance of expression of autosomal recessive disorder is rare, except in consanguinous marriages. When a phenylketonuric father gets married to his first cousin, the chance of the affected child is about 1 in 12, whereas in marriage with unrelated person the chance is about 1 in 10, 000.
(4) In sex-linked recessive disorder (say, haemophilia) when a boy is affected, his healthy mother should be a carrier and 50% of his sister are carrier of the disease. The carrier detection is a important task of the genetic counsellor. When an X-linked affected male (say, partial colour blindness) begets children, all daughters are carriers and all sons are normal.
(5) Sometimes individual seeks advice whether he will be affected by diabetes mellitus, since both of his parents are suffering from diabetes (autosomal recessive disorder.)
In such event, apart from blood glucose analysis of the individual, the anti-islet cells antibody titre of his serum will provide information whether he is pre-diabetic. He is then advised to follow dietary restriction.
Detection of Carriers:
The carriers may be detected by the following methods:-
Biochemical tests:
(1) Low level of catalase in acatalasia;
(2) Elevated serum level of creatin kinase in Duchenne's muscular dystrophy;
(3) Reduces factor VIII in haemophilia A;
(4) Reduced factor IX in haemophilia B;
(5) Reduced erythrocyte glucose-6- phospate in G-6-PD deficiency.
Amniocentasis:
(1) Prenatal determination of foetal sex by sex-chromatin study;
(2) Lecithin-sphingomyelin ratio of amniotic fluid for detection of foetal lung maturity;
(3) Alpha-fetoprotein level of amniotic fluid for detection of anencephaly and open spina bifida.
Foetoscope:
Use of foetoscope for collecting foetal blood from umbilical vessels helps prenatal diagnosis of sickle-cell anaemia and beta thalassaemia.