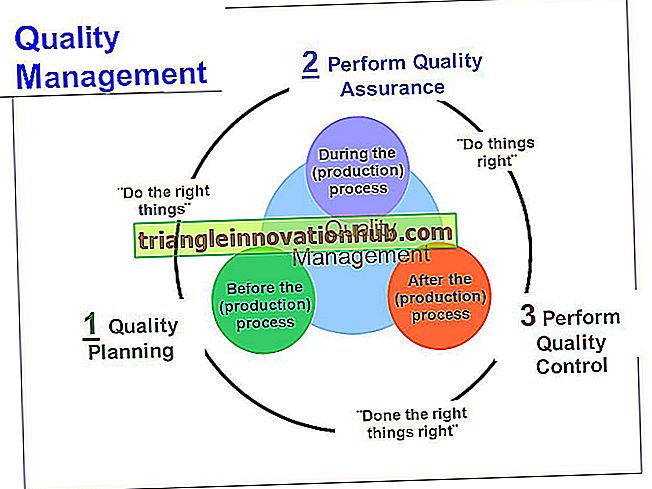
Stany kliniczne wynikające z reakcji nadwrażliwości typu II
Reakcja typu II jest znaczącym mechanizmem uszkodzenia tkanek w wielu chorobach autoimmunologicznych i innych stanach (takich jak niekompatybilna transfuzja krwi i hiperostre odrzucenie przeszczepu).
Reakcje nadwrażliwości typu II na krwinek czerwonych i płytki krwi:
Niezgodne transfuzje krwi:
System grup krwi ABO był pierwszym rozpoznanym systemem grup krwi. Grupa krwi ABO jest najważniejszym układem grupy krwi w transfuzji krwi. Istnieją cztery główne grupy krwi zwane A, B, AB i O, oparte na obecności lub nieobecności antygenu A i antygenu B na powierzchni czerwonych krwinek. Antygeny A i B są antygenami węglowodanowymi obecnymi na powierzchni czerwonych krwinek. Gen dla antygenów A i B jest obecny w chromosomie 9p i są one wyrażane w sposób współrzędny mendelski.
Przeciwciała przeciwko antygenom grupy A i B krwi występują naturalnie i należą do klasy IgM.
ja. Osobnik grupy A ma przeciwciała anty-B.
ii. Osobnik z grupy B ma przeciwciała anty-A.
iii. Grupa osobników AB nie ma przeciwciał anty-A i anty-B.
iv. Grupa O ma przeciwciała anty-A i anty-B.
Osoba, która oddaje krew, nazywa się "dawcą", a osoba, która otrzymuje krew, nazywa się "biorcą".

Ryc. 16.2: Mechanizm nadwrażliwości typu II.
Opsoniczna aktywność przeciwciał i regionów C3b: Fab przeciwciała wiąże się z antygenem na powierzchni komórki docelowej i inicjuje aktywację klasycznego szlaku dopełniacza. Fragment C3b utworzony podczas aktywacji dopełniacza spada na docelową błonę komórkową. Region Fc przeciwciała związanego z antygenem i C3b wiąże się odpowiednio z receptorem Fc i receptorem C3b na powierzchni komórki efektorowej (takiej jak makrofagi). Tak więc komórka docelowa jest połączona z komórką efektorową przez przeciwciało i C3b. Pseudopods komórek efektorowych otaczają komórkę docelową, przeciwciało i kompleks C3b i pochłaniają kompleks. Wewnątrz komórki efektorowej kompleks pochłonięty zostaje zniszczony
Odbiorca należący do grupy krwi A ma we krwi naturalnie przeciwciała anty-B. Jeśli zostanie mu podana krew grupy B / AB, region Fab przeciwciał anty-B (we krwi biorcy) będzie wiązał się z antygenem B na krwinkach czerwonych (transfuzji krwi B / AB).
↓
Region Fc przeciwciała anty-B związanego z RBC aktywuje klasyczny szlak dopełniacza.
↓
Aktywacja klasycznego szlaku dopełniacza prowadzi do lizy przetoczonych czerwonych krwinek i wywołuje reakcje transfuzji (takie jak spadek ciśnienia krwi, gorączka, uczucie ucisku w klatce piersiowej, nudności, wymioty).
Odbiorca grupy B ma przeciwciała anty-A i dlatego reaguje, jeśli krew A / AB jest transfuzowana do niego (Tabela 16.1). Grupa krwi O osobnika ma zarówno przeciwciała anty-A, jak i anty-B. Dlatego odbiorca grupy O będzie reagował z RBC od dawców A / B / AB.
Osobnik z grupy AB nie ma przeciwciał przeciwko antygenom A i B. Dlatego grupę osobników AB można transfundować z grupami krwi A / B / AB / O, a zatem osoby z grupą krwi AB są nazywane uniwersalnymi biorcami.
Tabela 16.1: Zgodni dawcy krwi i biorcy:
Odbiorca grupy krwi | Dawca O | Dawca ZA | Dawca b | Dawca AB |
O | - | + | + | + |
ZA | - | - | + | + |
b | - | + | - | + |
AB | - | - | - | - |
- Bez aglutynacji
+ Aglutynacja
RBC pojedynczego osobnika grupy O nie mają antygenu A i antygenu B na ich powierzchni. Dlatego też grupy RBC grupy O nie reagują z przeciwciałami anty-A i anty-B obecnymi w grupie A, grupie B lub grupie AB. Dlatego krew grupy O może być bezpiecznie transfuzowana do osób z grupy A / B / AB, a zatem osoby z grupy O są nazywane uniwersalnymi dawcami.
Choroba hemolityczna noworodków z powodu braku porównywalności Rh:
Obok systemu grup krwi ABO. System Rhesus (Rh) jest najważniejszym układem grupy krwi. System Rh został zademonstrowany przez Landsteinera i Weivera w 1940 r. Ich eksperyment polegał na wytwarzaniu przeciwciał przeciwko RBC małpy Rhesus u królików i świnek morskich. Naukowcy odkryli, że przeciwciała przeciwko RBCs małp Rhesus również aglutynują RBC 85 procent populacji ludzkiej.
Jeśli RBC osobnika były zlepione przez antyserum z RBC małpy Rhesus, osobnik ten miał czynnik Rhesus w swoich RBC (tj. Rh dodatni). Jeśli pojedyncze RBC nie były zlepione przez surowicę odpornościową RBC małpy Rhesus, osobnik nie posiadał czynnika Rh (tj. Rh ujemny). Teraz wiadomo, że system Rh jest złożony, a nasze obecne rozumienie opiera się na systemie Fishera.
Antygeny Rh znajdują się na błonie komórkowej RBC o 30 do 32 kDa. Antygen Rh nie ma określonej funkcji. W systemie Rh znajduje się około 40 różnych antygenów. Spośród nich pięć determinant antygenowych (zwanych D, E, e, C i c) jest bardzo powszechnych w populacji.
Osoby z antygenem D są nazywane Rh 'dodatnie, natomiast osoby bez antygenu D są nazywane Rh' ujemne. Układ antygenu D Rh jest silnym antygenem, a zatem antygen D indukuje silne odpowiedzi immunologiczne.
Gen Rh jest dominującym genem. W związku z tym dziecko Rh-dodatniego ojca lub Rh-dodatniej matki jest zawsze Rh pozytywne, niezależnie od statusu Rh drugiego partnera.
Dziecko Rh-dodatniego ojca i matki Rh-Rh jest Rh dodatnie. Rh dodatni płód w macicy matki Rh ujemnej nie powoduje żadnych oczywistych problemów dla matki, ale płód w macicy może rozwinąć się chorobę zwaną chorobą hemolityczną noworodka (HDN).
Płód Rh-dodatniego ojca i Rh-ujemnej matki będzie Rh dodatni. W czasie ciąży Rh dodatnia krew płodowa może wejść w krążenie matki Rh Rh.
↓
Antygeny Rh na płodowych RBC działają jako obcy antygen i indukują wytwarzanie przeciwciał anty-Rh u matki.
↓
Ponieważ wytworzone przeciwciała anty-Rh należą do klasy IgG, mogą przenikać przez łożysko i wchodzić w krążenie płodowe.
↓
Przeciwciała anty-Rh (od matki) wiążą się z antygenami Rh na płodowych RBC i hemolizują płodowe RBC.
Zniszczenie RBC nazywa się hemolizą. Stąd choroba nazywana jest chorobą hemolityczną noworodka (HDN). Hemoliza płodu RBC prowadzi do żółtaczki i anemii u płodu. Z powodu hemolizy wzrasta produkcja nowych czerwonych krwinek.
Zwiększenie produkcji erytrocytów jest znane jako erythroblastosis i dlatego choroba ta jest również znana jako erythroblastosis fetalis. Ponieważ Rh dodatnia krew płodu wchodzi w matkę Rh ujemną, stan ten jest również określany jako niezgodność Rh. Pierwsze dziecko, które urodziło się z niekompatybilną matką Rh, jest zwykle normalne i nie ma wpływu na HDN. Podczas gdy drugie i kolejne dzieci matki niekompatybilnej Rh rozwijają HDN.
Jaki jest mechanizm, który HDN wpływa na drugie i kolejne dzieci, podczas gdy pierwsze dziecko pozostaje nietknięte?
U ciężarnej krew płodowa oddzielana jest od krwi matki przez warstwę komórek zwanych trofoblastami w łożysku. W momencie porodu łożysko oddziela się od ściany macicy, co powoduje wnikanie niewielkiej ilości krwi od płodu do krążenia matki. Płodowe, Rh dodatnie RBC wchodzące do matki indukują wytwarzanie przeciwciał przeciwko antygenowi Rh.
Od pierwszego wejścia krwi płodowej do krążenia matki zwykle występuje w momencie porodu, pierwsze dziecko nie jest pod wpływem przeciwciał Rh. (Rozpoczęcie produkcji przeciwciał po wprowadzeniu płodowych krwinek czerwonych zajmuje wiele dni

Ryc. 16.3: Niedokrwistość hemolityczna wywoływana przez lek.
Lek lub jego metabolit może zostać zaadsorbowany na powierzchni RBC. Przeciwciała utworzone przeciwko metabolitowi leku / leku wiążą się z metabolitem lek / lek zaadsorbowanym na błonie RBC. Wiązanie antygen-przeciwciało prowadzi do aktywacji klasycznego szlaku dopełniacza.
Kompleksy atakujące błonę utworzone podczas aktywacji dopełniacza uderzają pory w błonę RBC i powodują lizę pierwszego dziecka RBC.) Podczas gdy drugi i kolejny płód są dotknięte przez przeciwciała Rh, ponieważ przeciwciała są obecne w matce nawet przed poczęciem. drugie dziecko.
Podczas drugiej i następnej ciąży do krążenia matki mogą dostać się małe ilości krwi płodowej. Antygeny Rh w płodowych RBC aktywują limfocyty B pamięci anty-Rh prowadzące do wytwarzania przeciwciał anty-Rh klasy IgG. Przeciwciało anty-Rh IgG wytwarzane przez matkę przechodzi przez łożysko i wchodzi w krążenie płodowe. Przeciwciała anty-Rh wiążą się z antygenem Rh na płodowych RBC i hemolizują RBC, powodując HDN.
Jednak rzadko pierwsze dziecko może również zostać dotknięte:
ja. Jeśli krew płodu wchodzi w krążenie matki na kilka miesięcy przed porodem, lub
ii. Matka miała już przeciwciała przeciwko antygenowi Rh, które mogą pojawić się z powodu przetoczenia krwi Rh-n przed nią lub w jej trakcie. Dlatego bezwzględnie konieczne jest sprawdzenie wszystkich kobiet w ciąży pod kątem ich statusu Rh. Jeśli spodziewana jest niezgodność Rh u kobiet w ciąży, należy sprawdzić jej poziom przeciwciał w surowicy na antygen Rh.
Jeżeli w surowicy obecne są przeciwciała, ilość przeciwciał powinna być okresowo sprawdzana. Jeśli występuje szybkie podniesienie poziomu przeciwciał lub poziom przeciwciał przekracza 2 xg / ml, należy wykonać amniocentezę, aby wykryć obecność hemolizy u płodu. Transfuzję krwi można podać płodowi w macicy, znaną jako transfuzja domaciczna.
Jaki jest główny mechanizm lizy krwinek czerwonych przez przeciwciała anty-Rh?

Ryc. 16.4: Mechanizm niszczenia płytek krwi podczas infekcji wirusowej.
Podczas infekcji wirusowej wytwarzane są przeciwciała przeciwko wirusom. Przeciwciała wirusowe wiążą się z wirusami w krążeniu i tworzą kompleksy antygen-przeciwciało. Przeciwciało związane z wirusem wiąże się z receptorem Fc na płytce krwi poprzez region Fc, region Fc przeciwciała związanego z wirusem również inicjuje aktywację układu dopełniacza. Kompleksy atakujące błonę tworzą się podczas perforacji otworów aktywujących dopełniacz na błonie płytki i poddają lizie płytki.
Przeciwciała IgG związane z antygenem Rh wiążą się z receptorami Fc na makrofagach w śledzionie i wątrobie. Makrofagi w śledzionie i wątrobie pochłaniają kompleks przeciwciało-RBC i niszczą RBC. Determinanty antygenowe Rh są rozmieszczone daleko na powierzchni krwinek czerwonych.
Chociaż klasa IgG przeciwciał anty-Rh wiąże się z antygenami Rh na powierzchni RBC, nie mogą one wiązać się z Clq, ponieważ przeciwciała związane z Rh są oddalone od siebie. (Wiązanie Clq wymaga obecności dwóch blisko położonych przeciwciał IgG związanych z antygenem, dlatego też nie występuje aktywacja klasycznego szlaku dopełniacza, dlatego też liza krwinek czerwonych przez lityczne składniki dopełniacza może nie być skutecznym sposobem hemolizy w HDN.
Zapobieganie występowaniu HDN w przyszłych ciążach:
Niezbędne jest podawanie przeciwciał anty-Rh-D matce matki Rh bezpośrednio po urodzeniu dziecka Rh dodatniego. Dokładny mechanizm działania podawanych przeciwciał anty-Rh-D nie jest znany. Uważa się, że przeciwciała anty-Rh-D pokrywają Rh-dodatnie płodowe RBC płodu i pośredniczą w ich usuwaniu, zanim będą mogły stymulować odpowiedź przeciwciał.
Wstrzyknięcie przeciwciała anty-Rh-D jest podawane wszystkim Rh ujemnym kobietom z Rh-i-płodem po porodzie, aborcji i każdej procedurze, która może indukować krwawienie przez łożysko (takie jak amniopunkcja) lub po przypadkowej transfuzji krwi Rh.
Oprócz niekompatybilności Rh, niekompatybilność ABO między matką a płodem może również wywoływać chorobę hemolityczną noworodka. Jednak takie choroby hemolityczne mają łagodny charakter. U płodu z grupy A lub B u matki matki może rozwinąć się choroba hemolityczna noworodka.
Autoimmunologiczna niedokrwistość hemolityczna:
Autoprzeciwciała to przeciwciała utworzone przeciwko własnym antygenom gospodarza (tj. Antygenom własnym). W stanach zwanych autoimmunologicznymi niedokrwistościami hemolitycznymi powstają przeciwciała przeciwko własnym antygenom błon komórkowych czerwonych krwinek gospodarza, które prowadzą do lizy czerwonych krwinek.
Wiązanie autoprzeciwciał z krwinkami czerwonymi powoduje zniszczenie czerwonych krwinek za pomocą następujących mechanizmów:
za. Aktywacja klasycznego szlaku dopełniacza. Kompleksy atakujące błonę utworzone podczas aktywacji dopełniacza lizują RBC.
b. Aktywacja klasycznego szlaku dopełniacza powoduje odkładanie się składników C3b na powierzchni RBC. Makrofagi śledziony mają receptory dla C3b. C3b na RBC wiąże się z receptorami C3b na makrofagach i w konsekwencji kompleksy C3b-RBC są pochłaniane przez makrofagi i niszczone (Fig. 16.2).
do. Region Fc autoprzeciwciała związanego z RBC wiąże się z receptorem Fc na makrofagach w śledzionie. W konsekwencji makrofag pochłania kompleks autoprzeciwciał RBC i niszczy RBC (ryc. 16.2).
Wywołana lekiem niedokrwienna niedokrwistość hemolityczna:
Podawanie leków może wywoływać niedokrwistość hemolityczną immunologiczną, chociaż takie stany są rzadkie. Istnieje wiele mechanizmów, dzięki którym leki mogą prowadzić do hemolizy immunologicznej.
za. Lek lub jego produkt metaboliczny może zostać zaadsorbowany na błonie komórkowej czerwonej (ryc. 16.3). Jeśli przeciwciała zostaną utworzone przeciwko lekowi, przeciwciało będzie wiązało się z lekiem zaadsorbowanym na krwinkach czerwonych i prowadzi do aktywacji dopełniacza. Lityczne składniki dopełniacza spadają na RBC i rozsyłają RBC.
b. Leki mogą działać jak hapteny, wiążąc się z białkami błonowymi RBC. W konsekwencji przeciwciała powstają przeciwko kompleksowi leków RBC.
Przeciwciała wiążą się z kompleksem antygen-lek błonowy RBC i prowadzą do lizy erytrocytów przez:
ja. Klasyczna aktywacja szlaku dopełniacza, oraz
ii. Poprzez fagocytozę kompleksu RBC-lek z udziałem receptora Fc przez makrofagi w śledzionie (np. Penicylina, chinina i chinidyna).
Małopłytkowość autoimmunologiczna:
Płytki krwi (trombocyty) są niezbędne do krzepnięcia krwi. Jeśli dojdzie do zniszczenia płytek krwi, powodując drastyczne zmniejszenie liczby płytek krwi, wpłynie to na krzepnięcie krwi. W konsekwencji pacjent będzie krwawił z wielu części ciała.
Idiopatyczna plamica małopłytkowa jest stanem klinicznym, w którym płytki krwi są niszczone przez mechanizm immunologiczny (trombocytopenia oznacza mniejszą liczbę płytek krwi, plamica oznacza wynaczynienie krwinek czerwonych w skórze). Ten stan występuje u wielu dzieci wracających do zdrowia po gorączce wirusowej lub chorobie górnych dróg oddechowych.
Płytki krwi mogą zostać zniszczone przez następujące mechanizmy:
ja. Podczas infekcji wirusowych powstają przeciwciała przeciwko wirusom, a przeciwciała wiążą się z wirusami. Kompleks wirus-przeciwciało wiąże się z receptorami Fc (przez region Fc przeciwciała związanego z wirusem) na błonach płytek krwi. Konsekwentna aktywacja klasycznego szlaku dopełniacza powoduje lizę płytek (ryc. 16.4).
ii. Przeciwciała produkowane przeciwko wirusowi mogą krzyżowo reagować z błonami płytkowymi (z powodu podobieństwa antygenowego, które może istnieć między wirusem a płytką krwi). Konsekwentna aktywacja dopełniacza lub fagocytoza za pośrednictwem receptora Fc przez makrofagi powoduje zniszczenie płytek krwi.
Leki mogą także wywoływać małopłytkowość poprzez immunologiczne niszczenie płytek krwi. Mechanizmy odpornościowe niszczenia płytek krwi podczas terapii lekowej są takie same, jak opisano w przypadku niszczenia czerwonych krwinek wywołanego przez leki. (Na przykład, Sulfathiazole, nowobiocyna, digitoksyna i metylodopa są niektórymi lekami, które mogą powodować zniszczenie płytek krwi za pośrednictwem układu odpornościowego).
Reakcja typu na antygeny tkankowe:
ja. Choroba błony podstawnej kłębuszków
ii. Pemphigus vulgaris
iii. Pemfigoid pęcherzowy
Choroba błony podstawnej kłębuszków (zespół Goodpasture'a):
W kłębuszkowej błonie podstawnej powstają autoprzeciwciała przeciwko kłębuszkowej błonie podstawnej (GBM). Autoprzeciwciała wiążą się z GBM i prowadzą do zniszczenia GBM, powodując chorobę nerek.
GBM składa się z kolagenu typu IV, lamininy, fibronektyny, proteoglikanów i entaktyny. Epitop w łańcuchu α3 kolagenu typu IV jest antygenem, z którym wiążą się przeciwciała GBM.
Przeciwciało anty-GBM wiąże się z błoną podstawną kłębuszkową i inicjuje aktywację klasycznego szlaku dopełniacza. C3a i C5a utworzone podczas aktywacji dopełniacza przyciągają neutrofile do miejsca odkładania przeciwciała w kłębuszku nerkowym.
↓
Neutrofile wiążą się z regionem Fc przeciwciała związanego z antygenem GBM, jak również C3b, odpowiednio poprzez ich receptory Fc i C3b, a neutrofile są aktywowane. W przeciwieństwie do drobnoustrojów błona podstawna nie może zostać pochłonięta przez neutrofile. W związku z tym neutrofile wylewają swoją zawartość komórkową ponad GBM, a zawartość uszkadza GBM prowadząc do niewydolności nerek.
Badania mikroskopowe immunofluorescencji wykazują liniowe odkładanie przeciwciał wzdłuż błony podstawnej kłębuszkowej (w chorobie nerek, w której pośredniczy receptor III typu III, występuje punktowe odkładanie kompleksów immunologicznych). Przeciwciała zazwyczaj należą do klasy IgG z dominującą podklasą IgGl. Często zdarza się, że zdeponowano również Clq i C3.
Istnieje podobieństwo antygenowe między nerkowym GBM a pęcherzykową błoną podstawną płuc. Dlatego przeciwciała GBM wiążą się również z pęcherzykiem płucnym w błonie płucnej, prowadząc do uszkodzenia pęcherzyków za pośrednictwem dopełniacza, a pacjent cierpi z powodu krwioplucia (hemoptysis oznacza kaszel krwi).
Obserwuje się związek między zakażeniem górnych dróg oddechowych a nawrotami choroby, w której pośredniczy przeciwciało anty-GBM. Jednak patogeneza nawrotu choroby GBM jest nieznana.
Leki immunosupresyjne (takie jak kortykosteroidy i cyklofosfamid) stosuje się w celu zmniejszenia wytwarzania autoprzeciwciał. Plazmaferezę można przeprowadzić w celu usunięcia autoprzeciwciał w krążeniu. W leczeniu niewydolności nerek może być wymagana dializa nerkowa i przeszczep nerki.
Pęcherzyca Wulgaris
(Pemphigus oznacza pęcherze, Vulgaris oznacza często):
Pemphigus vulgaris jest chorobą autoimmunologiczną skóry spowodowaną reakcją nadwrażliwości typu II, w której pośredniczą autoprzeciwciała. W pemphigus vulgaris komórki skóry oddzielają się od siebie, a uszkodzone pęcherze skórne są niszczone.
Desmosome jest spójnym elementem między komórkami naskórka. Desmoglin-3 (członek rodziny adhezyjnych cząsteczek komórek kadheryny) jest składnikiem białkowym desmosomu. Desmoglin-3 łączy ze sobą komórki skóry i inne nabłonkowe komórki. U pemphigus vulgaris wytwarzane jest autoprzeciwciało do desmogin-3, które wiąże się z desmoglinem-3 i prowadzi do tworzenia pęcherzy w skórze i błonach śluzowych. Przeciwciała przeciwko desmoglin-3 w surowicy nazywa się "przeciwciałami pęcherzycowymi".
Na skórze i błonach rozwijają się śródnabłonkowe, akantolityczne pęcherzyki i pęcherze (oddzielenie komórek naskórka od siebie nazywa się akantolizą). Badania immunofluorescencyjne wykazują międzykomórkową dystrybucję osadzania się IgG w skórze. Składniki dopełniające są również widoczne w skórze. (Jednak rola dopełniacza w immunopatogenezie nie jest jednoznaczna, ponieważ przeciwciała IgG z pęcherzyca zwykłego należą do podklasy IgG4, która nie aktywuje układu dopełniacza).
Pęcherzyca zwykła jest często spotykana u Żydów aszkenazyjskich i ma silny związek z HLA-DR4 i HLA-DQ3.
Niemowlęta urodzone przez matki z pęcherzycą zwykłą wykazują pęcherze na skórze przez okres przejściowy w okresie noworodkowym, co sugeruje, że choroba jest spowodowana przez przeciwciała IgG (które przenikają przez łożysko i docierają do płodu). Gdy IgG z pemphigus vulgaris pacjenta wstrzykuje się noworodkom myszy, myszy rozwijają pęcherze.
Choroba jest śmiertelna, jeśli nie jest leczona. Leki immunosupresyjne stosuje się w leczeniu stanu.
Pemfigoid pęcherzowy:
Pemfigoid pęcherzowy jest chorobą pęcherzykową pacjentów w podeszłym wieku. Napięte, podskórne pęcherze powstają na wewnętrznych udach i brzuchu. U 50% pacjentów obserwuje się zwiększoną liczbę eozynofilów i podwyższone stężenie IgE w surowicy. Bezpośrednie badania immunofluorescencyjne biopsji skóry wykazują liniowe i homogenne odkładanie się immunoglobulin i C3 w błonie podstawnej pod naskórkiem.
70 procent pacjentów ma krążące przeciwciała do strefy błony podstawnej skóry. Sugeruje się, że bulla rozwija się z powodu interakcji między antygenem w błonie podstawnej, przeciwciałem i dopełniaczem w reakcji nadwrażliwości typu II.
Reakcje typu II za pośrednictwem autoprzeciwciał antyreceptorowych:
Jak opisano powyżej, cytotoksyczność jest najczęstszą konsekwencją komórkowej reakcji autoprzeciwciała. Jednak nie zawsze tak jest. Niektóre choroby (takie jak myasthenia gravis i choroba Gravesa-Basedowa) wynikają z niecytotoksycznych interakcji między receptorami powierzchni komórki a autoprzeciwciałami przeciw receptorowi.
Myasthenia Gravis:
Myasthenia gravis jest zaburzeniem transmisji nerwowo-mięśniowej, a pacjenci cierpią na skrajne osłabienie mięśni. Choroba ta jest związana z obecnością autoprzeciwciał wobec receptorów acetylocholiny na błonie komórkowej mięśni w złączu nerwowo-mięśniowym.
Immunolog uodpornił króliki na oczyszczone receptory acetylocholinowe w celu wytworzenia przeciwciał przeciwko receptorom acetylocholiny. Ku swojemu zaskoczeniu, uodpornione króliki rozwinęły uszy. Dyskietki przypominają opadające powieki (opadanie powiek), które występują w myasthenia gravis u ludzi. Później wykazano, że pacjenci z miastenią istotnie mają przeciwciała przeciwko receptorom acetylocholiny.
Impuls nerwowy powoduje kontakt mięśnia. Impuls nerwowy powoduje uwolnienie acetylocholiny z zakończeń nerwowych w połączeniu nerwowo-mięśniowym (ryc. 16.5). Acetylocholina dyfunduje przez połączenie nerwowo-mięśniowe i wiąże się z receptorami acetylocholiny na błonie komórkowej mięśni, prowadząc do skurczu mięśnia. Acetylocholina ulega szybkiemu zniszczeniu przez enzym zwany esterazą acetylocholinową.
W miastenii rzekomej nie stwierdza się defektu w impulsie nerwowym lub wydzielaniu acetylocholiny. Autoprzeciwciała przeciw receptorowi acetylocholiny wiążą się z receptorami acetylocholiny na błonach komórek mięśniowych i zakłócają wiązanie acetylocholiny z receptorami.
Autoprzeciwciała przeciwko receptorowi acetylocholiny zmniejszają liczbę receptorów acetylocholiny na błonie komórkowej mięśni (ryc. 16.5).
ja. Przeciwciała wiążą się z sąsiednimi receptorami i sieciują receptory. W konsekwencji, kompleksy receptor-przeciwciało są internalizowane do komórki mięśniowej, przy czym kompleksy są niszczone. Ten mechanizm zmniejsza liczbę receptorów acetylocholiny na błonie komórkowej mięśni.
ii. Wiązanie się przeciwciała z receptorami prowadzi do mediowanych dopełnianiem uszkodzeń receptorów.
iii. Przeciwciała wiążą się z receptorami i zakłócają wiązanie acetylocholiny z receptorami.
Acetylocholina uwolniona podczas impulsu nerwowego może nie wiązać się z żadnymi receptorami lub może wiązać się z bardzo niewielką liczbą dostępnych receptorów. Rezultatem jest to, że aktywacja mięśni jest poważnie zakłócona. Pacjent odczuwa osłabienie mięśni i nie jest w stanie unieść nawet powiek (a więc opada powieki).
Pirydostygmina hamuje enzym esterazę acetylocholinową (która normalnie inaktywuje acetylocholinę). Podawanie pirydostygminy przedłuża biologiczny okres półtrwania acetylocholiny, a zatem stosuje się ją w leczeniu myasthenia gravis.

Ryciny od 16.5A do D: Schemat połączenia nerwowo-mięśniowego w miastenii ciężkiej. (A i B) Normalne połączenie nerwowo-mięśniowe:
(A) Zakończenie nerwu ma acetylocholinę, a błona mięśniowa ma wiele receptorów dla acetylocholiny, (B) Podczas impulsu nerwowego acetylocholina jest uwalniana z zakończenia nerwowego. Uwolniona acetylocholina wiąże się z receptorami acetylocholiny na błonie komórkowej mięśni i prowadzi do kurczenia się komórek mięśniowych.
Połączenie nerwowo-mięśniowe w myasthenia gravis (C i D): (C) W miastenii ciężkiej autoprzeciwciało receptora acetylocholiny wiąże się z receptorem acetylocholinowym i prowadzi do internalizacji kompleksu autoprzeciwciało-acetylocholina do komórki mięśniowej, w którym są niszczone. W ten sposób zmniejsza się liczba receptorów acetylocholiny na powierzchni błony komórek mięśniowych i (D) Autoprzeciwciało z receptorem acetylocholinowym wiąże się z receptorem acetylocholiny na błonie komórkowej mięśni i zakłóca wiązanie acetylocholiny z receptorami. W konsekwencji wpływa to na skurcz komórek mięśniowych
Autoprzeciwciała przeciwko receptorom acetylocholiny należą do klasy IgG. Dlatego autoprzeciwciała przeciwko receptorowi acetylocholiny IgG u kobiet w ciąży mogą przenikać przez łożysko i wchodzić w krążenie płodowe. W konsekwencji nowo narodzone niemowlęta matek z miastenią rzekomą wykazują objawy myasthenia gravis przy urodzeniu. Jednak objawy trwają tylko od jednego do dwóch tygodni.
U niemowląt przeciwciała wiążą się z receptorami acetylocholinowymi na błonach komórek mięśniowych, a kompleksy receptor-przeciwciało acetylocholinowe są internalizowane do komórek mięśniowych i niszczone. W ciągu 10 do 15 dni wszystkie macierzyste przeciwciała receptora acetylocholinowego są usuwane z krążenia niemowlęcia, a objawy niemowlęcia znikają.
Choroba Gravesa-Basedowa:
Choroba Gravesa-Basedowa jest zaburzeniem autoimmunologicznym, które dotyczy głównie tarczycy. W tym zaburzeniu pośredniczą autoprzeciwciała, które stymulują aktywność komórek tarczycy, prowadząc do nadmiernej produkcji hormonów tarczycy, które są odpowiedzialne za prezentację kliniczną.
Istnieją trzy kategorie przeciwciał przeciwtarczycowych, które zmieniają funkcje tarczycy. Immunoglobina hamująca wiązanie tarczycy (TBI) [znana również jako przeciwciało przeciw receptorowi hormonu stymulującego (TSH)] jest jednym z trzech przeciwciał przeciwtarczycowych. Normalnie hormon tarczycy (TSH) wydzielany przez przysadkę wiąże się z receptorem hormonu tarczycy (receptorem TSH) na tarczycy i stymuluje tarczycę do produkcji hormonów tarczycy. Hormony tarczycy we krwi działają na przysadkę mózgową i wysyłają sygnał ujemnego sprzężenia zwrotnego, co prowadzi do zmniejszenia wydzielania TSH. Tak więc poziom hormonów tarczycy utrzymuje się w normalnych granicach.
Wiązanie TBI z receptorem TSH prowadzi do ciągłej stymulacji tarczycy iw konsekwencji hormony tarczycy są wydzielane w dużych ilościach. Zwiększone poziomy hormonów tarczycy są odpowiedzialne za kliniczne objawy choroby Gravesa-Basedowa.
Reakcja typu II na przeszczepione narządy:
Hiperacute odrzucenie przeszczepu występuje, gdy biorca przeszczepu wstępnie wytworzył przeciwciała przeciwko antygenom przeszczepionym. Wytworzone przeciwciała przeciwko antygenom tkankowym mogły zostać wywołane przez wcześniejsze przetoczenie krwi lub wcześniejsze przeszczepy. Te wstępnie utworzone przeciwciała reagują z przeszczepionymi antygenami na komórkach przeszczepionych i indukują reakcje typu II. (Reakcja antygen-przeciwciało prowadzi do infiltracji neutrofili.
Neutrofile są łączone mostkowo z komórkami przeszczepionymi poprzez receptory Fc i C3b na neutrofilach. Neutrofile rozładowują swoje enzymy i toksyczne składniki na komórkach. W transplantacji nerki reakcja ta prowadzi do poważnego uszkodzenia naczyń włosowatych kłębuszków i ostatecznie zniszczony jest przeszczep. Ta reakcja zwykle występuje od kilku minut do 48 godzin po zakończeniu operacji przeszczepu.